
The Hedgehog (Hh) pathway is a developmental signaling egy for a variety of degenerative or ischemic disorders has also pathway that plays multiple roles during embryonic develop been proposed; however, the development of small-molecule ment and in adult tissues. Constitutive Hh signaling has been Hh agonists has received less attention. The goal of this review linked to the development and progression of several forms of is to highlight the recent evidence supporting the therapeutic cancer, and the application of small-molecule pathway inhibitors potential of Hh pathway activators and to provide a compretors as anticancer chemotherapeutics is well studied and clear hensive overview of small-molecule pathway agonists.
Introduction
In cells regulated by the Hedgehog (Hh) pathway, signal transmission is controlled through a cascade of events that ulti-mately results in a balance between activator (GliA) and repressor (GliR) forms of the glioma-associated oncogene (Gli) family of transcription factors. As this process has been extensively reviewed elsewhere, only a brief description of the key regulators is described herein. There are three Hh proteins [Sonic (SHh), Indian (IHh), and Desert (DHh)] that serve as ligands to initiate pathway signaling. In the absence of an Hh protein, Patched (Ptch), a 12-transmembrane domain protein located on the cell surface, suppresses the activity of Smoothened Agonist, a 7-transmembrane GPCR-like receptor primarily located in the membrane of intracellular vesicles . When Smo is inactive, Suppressor of Fused (Sufu), a negative regulator of Gli, forms a heteroprotein complex with Gli proteins in the cytosol. This complex promotes phosphorylation of the Gli proteins, leading to the generation of N-terminal trun cated forms, GliR, that act as repressors of Hh responsive genes. Binding of an Hh ligand to Ptch results in its internalization and abolishes its inhibition of Smo, which undergoes phosphorylation at its C terminus followed by reciprocal translocation to the plasma membrane of non-motile cilia . This process prevents the proteolytic processing of Gli proteins in the cytosol and results in the production of fulllength activator forms of Gli (GliA). Translocation of GliA to the nucleus stimulates transcription of Hh target genes, including GLI1 and PTCH1.
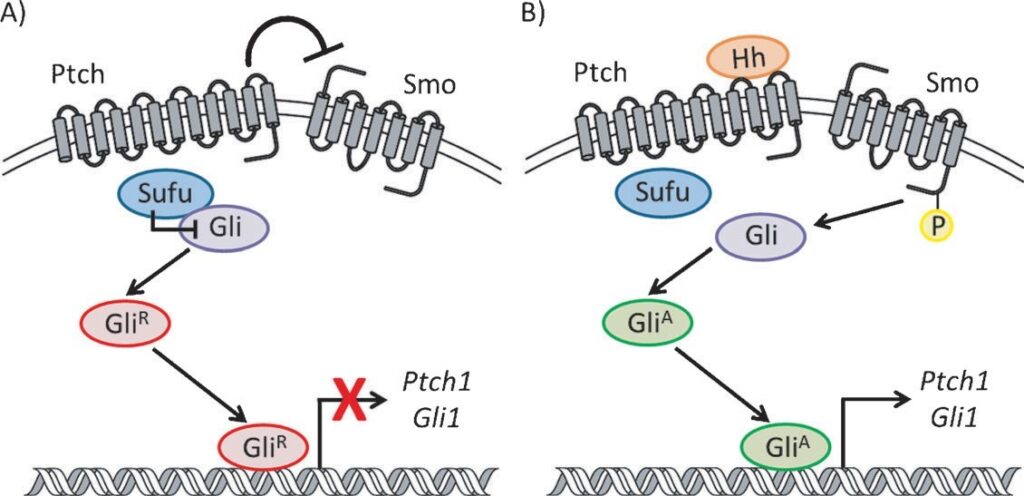
Figure 1. Generalized scheme for the Hh pathway signaling cascade.
Therapeutic Applications of Hh Pathway Agonists
While the use of small-molecule Hh pathway inhibitors as anticancer chemotherapeutics has been extensively studied, the therapeutic application of Hh pathway agonists has generated less interest. Recent years have seen an increase in publications describing the Hh pathway in the central and peripheral nervous systems, the process of bone formation, and the vasculature. Extensive reviews into the nature of Hh signaling in the development and maintenance of these tissues have been published elsewhere. This section will highlight key aspects of the Hh pathway in these systems and provide an overview as to how modulation of the pathway holds therapeutic potential within these cellular contexts.
Neurodegenerative disorders Hh signaling has been identified as an integral component for neural system patterning in both the central and peripheral nervous systems (CNS and PNS). In the CNS, SHh signaling regulates the specification of multiple neural cell types, including motor, dopaminergic, and basal forebrain cholinergic neurons. In addition, SHh stimulates proliferation of neural precursors. DHh appears to play an essential role in PNS development; however, evidence suggests it guides proper development and function of nerve sheaths through an indirect method in which DHh derived from Schwann cells stimulates the secretion of neurotrophic growth factors on perineurial cells. In addition to its role during neural system development, proper Hh signaling plays a crucial role in the adult nervous system; most notably, through regulation of neuronal stem cell populations and neuronal cell fate determination.
To date, most of the published data exploring activation of Hh signaling as a therapeutic strategy against CNS neurodegenerative disorders has focused on Parkinson’s disease (PD), a degenerative disorder characterized by the loss of dopaminergic neurons in the substantia nigra. Direct intrastriatal administration of SHh protected against drug-induced neurotoxicity and improved motor function and locomotor activity in multiple animal models of PD. In addition, adenoviral vector-mediated transfer of SHh and Gli1 was sufficient to protect dopaminergic neurons in the substantia nigra from the neurotoxin 6-hydroxydopamine (6-OHDA) in a similar rodent model of PD. SHh was required to generate midbrain dopaminergic neurons from embryonic stem cells derived from either neuroepithelial or anterior neural plate cells, and SHh has been used (along with fibroblast growth factor 8 and basic fibroblast growth factor) to generate dopaminergic neurons from adult human mesenchymal stem cells. SHh has also demonstrated neuroprotective effects in an in vitro model of amyotrophic lateral sclerosis following oxidative insult.
Induction of Hh signaling has been studied as a potential treatment for peripheral diabetic neuropathy and nerve injury. Diabetic neuropathy is a common and serious complication of diabetes in which long-term high blood sugar levels result in the progressive degeneration of peripheral nerves. In a rat model of induced diabetes, DHh mRNA was significantly reduced in peripheral nerves, and subcutaneous treatment with an SHh-IgG fusion protein restored nerve conduction velocities to non-diabetic levels. A similar in vivo study demonstrated the ability of SHh to improve nerve function with the additional effect of improving neural blood flow. The neuroprotective effect of SHh has also been demonstrated in in vivo models of acute nerve injury. SHh and DHh mRNA was significantly increased in a mouse nerve crush model, and antagonizing Hh signaling with anti-Hh monoclonal antibodies prolonged recovery time. By contrast, periodic injection of a modified form of recombinant SHh accelerated recovery from the nerve injury. Following crush injury to the sciatic nerve of adult rats, SHh and Gli1 mRNA was upregulated in adjacent Schwann cells, ultimately resulting in the stimulation of brain-derived neurotrophic factor (BDNF). Continuous administration of the Hh inhibitor cyclopamine (Cyc) suppressed both BDNF expression and motor neuron survival. Taken together, these studies indicate that upregulation of Hh signaling could promote survival, regeneration, and/or increased function of remaining neurons and could be a valid strategy for treating numerous neurodegenerative disorders.
Osteodegenerative disorders
Hh signaling is essential for guiding the proper proliferation and differentiation of osteoprogenitor cells during embryonic bone formation; activity that is primarily linked to IHh. IHh—/— mice demonstrate defects in endochondral bone patterning and fail to form osteoblasts, leading to stunted limb formation. These effects can either be through direct initiation of osteogenic differentiation in osteoblast progenitor cells or via an indirect mechanism in which IHh induces expression of other cellular factors, primarily, parathyroid hormone-related protein (PTHrP) that initiate bone formation. Evidence also points to a sequential series of steps in which IHh initiates the differentiation of skeletal precursor cells into osteoblasts, followed by canonical Wnt/b-catenin signaling to promote a terminal osteoblastic cell fate. Recent studies into the role of Hh signaling during adult bone homeostasis have demonstrated that pathway activation in this context is less clear cut. Selective upregulation of Hh signaling in mature osteoblasts led to excessive bone resorption and severe osteopenia, a precursor state to osteoporosis. By contrast, inhibition of the pathway in this model increased bone mass in aged mice. These data suggest that activation of Hh signaling has the potential to generate new bone growth and may hold potential as a treatment for degenerative bone diseases such as osteoporosis; however, detrimental side effects, including increased risk for osteoporosis, are possible with prolonged pathway activation.
Hair growth
SHh plays a vital role in hair follicle morphogenesis during normal skin development and continues an essential role in regulating follicular growth and cycling in adults. While not essential for hair follicle initiation during embryonic development, SHh is required for controlling growth and morphogenesis of the follicle past the hair germ stage. SHh—/— mice exhibit normal levels of hair placodes; however, dermal papillae do not form properly and follicular epithelium proliferation is impaired. The reduction in hair follicle papillae correlated with decreased expression of both Gli1 and Ptch1. In adults, SHh mediates the transition from the resting (telogen) to the growth stage (anagen) during the normal follicle cycle and anti-SHh antibodies prevented hair growth in adult mice. Taken together, these studies suggest activation of Hh signaling holds therapeutic potential in diseases characterized by decreased follicular proliferation and altered cycling (such as androgenetic alopecia).
Diabetes and obesity
Initial studies into the ability of Hh signaling to regulate metabolism demonstrated a conserved role in Drosophila and mammals in which Hh signaling inhibited fat formation. A genome-wide RNAi screen in adult Drosophila, designed to identify genes linked to obesity, identified the Hh signaling pathway as the top scoring pathway specific to the formation of fat. Interestingly, this study demonstrated that the pathway specifically blocks white adipogenesis, but has no effect on brown adipogenesis. Follow-up studies aimed at understanding the cellular mechanisms that govern Hh modulation of fat formation and cellular metabolism identified a non-canonical Smo-dependent signaling mechanism that rewires cellular metabolism. A Smo-Ca2+-Ampk axis, active in the primary cilia, rapidly induces Warburg-like metabolic reprogramming that is required for proper metabolic function. Activation of this axis in vivo induces insulin-dependent glucose uptake in both muscle and brown fat tissue, suggesting that Hh pathway agonists hold therapeutic potential for the treatment of diabetes and obesity.
Ischemia
Ischemic diseases are characterized by reduced blood flow to the tissues, resulting in a decrease in oxygen, glucose, and other nutrients required for proper cellular function. The most prominent forms of ischemia include ischemic heart disease, myocardial infarction, and stroke. A primary therapeutic strategy to treat ischemia is revascularization of the damaged or degenerative blood vessels. The Hh signaling pathway is integral during the development of the cardiovascular system and is sufficient to promote the formation of new coronary vessels in adults. As examples, mouse (Smo—/—) and zebrafish (SHh—/—) embryos lacking key pathway components exhibit defective vasculogenesis, and activation of the Hh pathway in the adult mouse promotes angiogenesis and vascularization of several different tissues. In a murine model of ischemia, recombinant SHh induced neovascularization by upregulating several angiogenic factors, an effect that was inhibited with the addition of an anti-SHh antibody. Within the context of myocardial infarction, direct administration of an SHh-expressing plasmid into the myocardium during infarct preserved myocardial function. Follow-up studies suggested that the gene therapy increased expression of SHh ligand and promoted the expression of various angiogenic and anti-apoptotic factors. In a murine stroke model, enhanced SHh was seen in hippocampal neural progenitor cells (NPCs) following insult, resulting in increased proliferation. These effects were enhanced with the exogenous addition of recombinant SHh and antagonized with Cyc. More recently, brain edema associated with cerebral ischemia was reduced following administration of SHh through a process identified as Smo-dependent.
Small-Molecule Hh Pathway Agonists
Sterol-based small-molecule agonists Endogenous sterols are known to play an integral role in regulating the Hh pathway and Smo signaling.
Following the autocatalytic cleavage from its precursor form, the Hh protein is covalently bound to a cholesterol moiety at its C terminus, and this modification is essential for full signaling activity in vivo. A recent study provided strong evidence that PTCH regulates the intracellular cholesterol concentration of Hh-dependent cells by contributing to cholesterol efflux, and that this activity may be a factor in preventing Smo accumulation at the plasma membrane. In addition, several exogenous sterols and sterol-like small molecules have been identified as potent modulators of Hh signaling. The secosteroid vitamin D3 and the steroidal natural product Cyc are potent inhibitors of Hh signaling that are currently being explored as anticancer agents. Both natural and synthetic oxysterols (OHCs), byproducts of cholesterol oxidation, have been shown to activate Hh signaling through direct interactions with Smo. Finally, several synthetic fluorinated glucocorticoids (GCs) were recently identified as activators and inhibitors of pathway signaling. With this in mind, this section of the review will focus on OHCs and GCs as Hh pathway agonists.
Endogenous oxysterols
Early studies on the physiologic effects of oxysterols (OHCs) in pluripotent mesenchymal stem cells and osteogenic progenitor cells (M2-10B4) focused on exploring the osteoinductive and anti-adipogenic properties of these compounds, effects that were subsequently linked to their ability to activate Hh signaling. A separate report, studying mechanisms through which the sterol biosynthetic pathway regulates Hh signaling, identified OHCs as potent Hh pathway agonists in medulloblastoma cells. Both of these early studies focused on evaluating naturally occurring OHCs with a single hydroxy moiety. OHCs with hydroxy substitutions on the side chain in close proximity to the tetracyclic core, including 20(S)-OHC (1, EC50 =0.1 mm) and 22(S)-OHC (2, EC50 = 0.2 mm), demonstrated potent pathway activation. Interestingly, orientation of the hydroxy was crucial for pathway activation as 22(R)-OHC was approximately 10-fold less active than 22(S)-OHC. OHCs in which the hydroxy moiety was located at the distal end of the side chain, such as 24-OHC (3) and 25-OHC (4), were modest pathway activators (EC50 = 3 mm and 1 mm, respectively). By contrast, OHCs in which a hydroxy moiety is only located on the tetracyclic core (7band 19-OHC, 5 and 6) have proven inactive, highlighting the importance of a hydroxy moiety in the side chain region of the OHC scaffold. Two endogenous OHCs that incorporate multiple substitutions, a ketone at the 7-position and a hydroxy in the side chain [7keto-25-OHC (7) and 7-keto-27-OHC (8)], have been recently identified as potent activators of the Hh signaling pathway.
Both 7 and 8 (10 mm) upregulated pathway signaling approximately 20-fold in Hh-dependent cell culture. It is important tonote that the structurally related oxysterol 7a-27-OHC did not activate Hh signaling, suggesting that proper orientation of the oxygen moiety in the 7-position is essential for pathway agonism.
Synthetic oxysterols
Following the identification of naturally occurring OHCs as Hh pathway activators, several research groups have synthesized and evaluated differentially hydroxylated OHC analogues to further explore the structure–activity relationships (SAR) for this class of Hh pathway agonists. The first group of researchers to undertake SAR studies for the OHC scaffold focused on analogues of 20(S)-OHC that incorporated either (1) various hydrophobic moieties in the side chain or (2) an additional hydroxy moiety at C-6. In general, OHCs with an aromatic side chain functionality were less active than those containing an aliphatic chain, and a free hydroxy at C-6 enhanced activity. The most active OHCs identified through these initial studies, Oxy34 (9) and Oxy49 (10), demonstrated potent Hh pathway activity as measured by their ability to upregulate alkaline phosphatase (ALP) activity in M2-10B4 cells, a well-characterized downstream effect of Hh signaling (EC50 =0.8 mm and 0.9 mm, respectively). Both Oxy34 and Oxy49 treatment (5 mm) induced overexpression of the Hh target genes GLI1 and PTCH1 in M2-10B4 cells followed by osteogenic differentiation of these cells into mature osteoblasts, indicating functional activation of Hh signaling. In addition, both OHCs induced new bone formation in a murine model of spinal fusion. Administration of 20 mg (via collagen sponge implant) of either Oxy34 or Oxy49 resulted in spinal fusion in each rat of the treatment group, whereas a 2 mg dose of Oxy34 induced spinal fusion in 50 % of rats dosed.
A second generation series of Oxy34 analogues has recently provided further input into the SAR for the side chain region of this scaffold. The most potent of these analogues incorporate an unbranched aliphatic side chain. Both n-hexyl (11) and n-heptyl (12) side chain analogues demonstrated potent Hh agonism (EC50 = 0.4 mm and 0.5 mm, respectively). Similar to the first generation OHCs, analogues with a bulky phenyl or pyridyl side chain demonstrated reduced potency (EC50 range between 2.5 mm and > 5 mm). In addition, modification of the C21 methyl to an ethyl decreased activity (EC50 = 2.9 mm), whereas larger substitutions (phenyl, i-hexyl) at this position completely abolished pathway activation. Interestingly, stability data obtained in human liver microsomes showed that the most active OHCs exhibited poor stability (< 5 % remaining at 1 h). Based on the rationale that metabolism of these analogues begins with C-22 hydroxylation, a focused series of C22, C-23 deuterated analogues of Oxy34 were synthesized and evaluated. The most active of these, the tetradeuterated analogue (13), activated Hh signaling at a level similar to Oxy34 (EC50 = 1.2 mm) and demonstrated enhanced stability (T1/2 > 120 min). Both 11 and 13 were evaluated for induced bone formation in the rat spinal fusion model (4 mgkg—1); however, only 11 demonstrated a significant increase in bone volume compared with Oxy34. These results suggest that the in vitro metabolic stability of this class of OHC Hh agonists does not provide a direct corollary to in vivo efficacy.
A separate series of OHCs with varying hydroxylation patterns in the side chain was recently synthesized and evaluated for their ability to activate Hh signaling. These compounds focused on OHCs with both single and multiple hydroxy substitutions in the side chain region. In addition, Hh pathway selectivity for this series of OHCs was evaluated by measuring their ability to activate signaling associated with the liver X receptor (LXR), a nuclear receptor that also binds and responds to various OHCs. The results from these studies provided important SARs for this series. First, none of the compounds that incorporate a C-25 hydroxy were selective for the Hh pathway. Second, analogues with a truncated side chain were less potent Hh pathway agonists. The most selective analogue from this series was 23(S)-OHC (14, EC50 = 0.54 mm, approximately 18-fold selective for Hh signaling). Further evaluation of 23(S)-OHC showed its ability to upregulate both early and late transcriptional markers of osteogenic differentiation in vitro, thus demonstrating functional activation of Hh signaling.
A recent publication, focused on determining the cellular mechanisms that regulate OHC activation of Hh signaling, identified the terminal alkyne nat-20(S)-yne (15) as a potent activator of Hh signaling. Nat-20(S)-yne was initially prepared as a synthetic intermediate to the bead-linked 20(S)-OHC but its evaluation identified it as a potent agonist of Hh signaling in vitro (EC50 ≈ 390 nm). Follow-up studies with this OHC have not been reported. Finally, a focused series of 20(S)and 20(R)-OHC analogues that incorporated modified alkyl side chains has been reported. SAR for this series demonstrated that, similar to previous reports, 20(R)-OHC analogues were unable to activate the Hh pathway. 20(S)-OHC analogues with shorter alkyl side chains (methyl, ethyl, and propyl) did not activate Hh signaling, whereas extended side chains [butyl (16) and pentyl (17)] significantly upregulated Hh signaling. While definitive EC50 values for these OHCs were not reported, the most potent analogues upregulated Hh signaling with approximate EC50 values of 1 mm [20(S)-OHC-Pent] and 10 mm [20(S)-OHC-Bu]. In addition, saturation of the 5,6-olefin in the pentyl analogue resulted in 20(S)-OHC-PentSat (18), which maintained the ability to activate Hh signaling (EC50 ≈ 10 mm). Finally, masking the 3bOH with a methoxy completely abolished activity, suggesting the free hydroxy moiety is essential for the potent pathway activation elicited by the OHC scaffold.
OHCs activate Hh signaling via Smo
As endogenous OHCs are the only natural metabolites that have been identified as Hh pathway agonists, there has been much interest in characterizing the mechanisms through which these compounds activate Hh signaling. Three recent studies have definitively identified that OHCs do, in fact, exert their agonist activities via direct binding to Smo. The first of these reports used the triazole-linked 20(S)-OHC beads (19) in a series of assays to demonstrate a direct binding interaction between 20(S)-OHC and Smo. Through a series of deletion and ligand affinity studies, the amine-linked 20(S)-OHC beads (20) shown in were used to localize OHC-Smo binding interactions to the extracellular cysteine-rich domain (CRD) on Smo. It is important to note that a structurally related cholesterol-like structure containing an amine in the side chain region (22-NHC, 21) bound to the OHC binding site on SmoCRD and inhibited Hh signaling, demonstrating that this binding site could potentially be targeted for the development of Hh pathway inhibitors. A related series of experiments also demonstrated that OHC agonists of Hh signaling exert their effects via direct binding to the SmoCRD and provided evidence that binding interactions between 20(S)-OHC and the SmoCRD are localized to “site 1” of the CRD, a hydrophobic pocket formed by side chain a helices. In addition, the modulatory effects of OHC binding to the SmoCRD are distinct from those related to inhibition of the pathway by the Cyc. Taken together, this data provides evidence that pathway signaling at the level of Smo can be modulated by steroidal small molecules at multiple binding sites.
Synthetic glucocorticoids
A high-content screen in U2OS cells stably expressing a b-arrestin2-GFP reporter identified several FDA-approved synthetic glucocorticoids (GCs) as Hh pathway agonists. In this model system, overexpression of SMO followed by treatment with a small-molecule SMO agonist results in aggregation of b-arrestin2-GFP in intracellular vesicles. Initially, four fluorinated GCs were identified as Smo agonists at the screening concentration (5 mm): fluticasone propionate, halcinonide, clobetasol, and fluocinonide . Follow-up studies in the screening assay demonstrated that fluticasone propionate (22, 99 nm), halcinonide (23, 1.1 mm), and clobetasol (24, 1.5 mm) dose-dependently upregulated Hh signaling with EC50 values below 5 mm, and a subsequent screen of a biased steroid library (1658 compounds) did not identify additional pathway agonists. Interestingly, each of these GCs were significantly more effective at upregulating Gli levels in Shh-LIGHT2 cells (EC50 = 0.3–1.8 nm).
Competitive binding experiments demonstrated that all three GCs displaced BODIPY-Cyc from Smo; however, maximal displacement was modest (20–25 %) in comparison to the small-molecule SMO agonist (SAG, 100 % displacement) at the highest concentrations evaluated (10 mm). The GCs were also evaluated for their ability to stimulate proliferation of cerebellar granular cell precursors (GCPs), a cell type that requires Hh/ Smo signaling for expansion and differentiation in vivo. Whereas treatment of GCPs with fluticasone propionate and clobetasol resulted in a modest increase in proliferation (fiveto sixfold), treatment with halcinonide increased proliferation 50fold and synergized with SHh to upregulate proliferation approximately 80-fold ciliary accumulation, and Hh pathway activation. First, a ketal bridging C-16 and C-17 was significantly more effective at inducing Smo ciliary accumulation than the free alcohols. This was most apparent when comparing the activities of triamcinolone acetonide (27, EC50 5 mM) and triamcinolone (28, EC50 ≈ 75 mM). Second, significant differences were seen between Subsequent to their initial identification as Hh agonists, an independent cell-based screen designed to identify small molecules that induce Smo ciliary accumulation identified several GCs as regulators of this process. The results from this screen and follow-up studies with these compounds provided interesting information with regards to the GC scaffold, Smofluocinolone acetonide (FA)-induced Smo ciliary accumulation and Hh pathway activation. Low-micromolar concentrations of FA (26, 10 mM) promoted ciliary accumulation; however, significantly higher concentrations (> 50 mM) elicited only a modest fourfold increase in Gli levels. FA displaces BODIPY-Cyc from Smo in a dose-dependent fashion (IC50 ≈ 50 mM) and, in contrast to the study described above, complete displacement occurred at concentrations ≥ 100 mM. Of note, two structurally-related GCs, budesonide (29) and ciclesonide (30), inhibited Hh signaling (IC50 ≈ 40 mM), suggesting that scaffolds of this type can be developed as both activators and inhibitors of Hh signaling.
Additional synthetic small-molecule Hh agonists SAG
A high-throughput screen of 140 000 small molecules performed in a C3H10T1/2 clone, stably expressing a plasmid containing a luciferase reporter downstream of a minimal Gli promoter, identified a family of biaryl benzothiophenes as Hh pathway agonists. The initial screening hit, Hh-Ag 1.1, demonstrated potent pathway activation with an EC50 value of 0.4 mm. In this report, data for several small-molecule agonists of this scaffold was disclosed (Hh-Ag 1.1–Hh-Ag 1.5). The structures of Hh-Ag 1.1, 1.2, and 1.3 (later renamed SAG for Smoothened agonist) were initially disclosed with the structure for Hh-Ag 1.5 disclosed in a separate report . Both Hh-Ag 1.2 and Hh-Ag 1.5 were potent activators of Hh signaling in the clonal C3H10T1/2 cell line (EC50 ≈ 10 nm and 1 nm, respectively). In addition, Hh-Ag 1.2 demonstrated potent pathway activation in primary neonatal cerebellar granule neuron (CGN) precursors in vitro, and SAG stimulated pathway signaling in vitro following oral administration. Preliminary studies designed to determine the mode of action for this class of compounds used a tritiated diazirine derivative of Hh-Ag 1.2 in photoaffinity labeling studies and showed that it bound exclusively to Smo. Similar studies with a photoaffinity-labeled analogue of SAG (PA-SAG, 34) demonstrated that this compound also directly binds Smo and localized its binding site to the Smo heptahelical bundle.
Since these initial studies, two detailed SAR reports in the scientific literature have provided insight into the design and structural requirements for pathway activation for this class of Hh pathway agonists. First, for the central 1,4-diaminocyclohexane moiety the trans orientation was critical for activity, and while the addition of a singular methyl group to the 4-position amine enhanced activity approximately 125-fold, larger substituents completely abolished activity. SAR for the 3chlorobenzothiophene group demonstrated that replacement of the chlorine with a methyl was tolerated; however, substitution with a hydrogen resulted in a complete loss of activity. In addition, removal of the distal phenyl ring resulted in an inactive analogue. A single fluorine substitution at either the 4-, 5-, or 7-position of the benzothiophene significantly enhanced activity of the scaffold, while fluorine and other substituents in the 6-position were detrimental to activity. Incorporating fluorines at both the 4and 7-positions resulted in an analogue that potently activated Hh signaling (35, EC50 = 2 nm). Finally, SAR for the biaryl region was explored. Optimal substitutions in this region included a methoxy in the 4-position of the proximal aryl ring, and incorporation of the heterocyclic 4-pyridyl moiety as the distal aromatic ring. Combining the optimized functionalities for each region into a single analogue provided 36 as a potent Hh pathway agonist (EC50 = 0.3 nm). Compound 36 also demonstrated a promising pharmacokinetic profile in mice and was able to penetrate the central nervous system, achieving a Cmax of 81 ngmL—1 and T1/2 of 4 h following oral administration (5 mgkg—1). To date, further in vitro and in vivo activity for 36 in Hh-related disease models has not been reported in the scientific literature.
The majority of studies that have explored the Hh activation of this class of compounds within a disease context have specifically focused on SAG. Combination treatment of mouse embryonic stem (ES) cells with SAG and retinoic acid induced their differentiation into spinal progenitor cells and, ultimately, functional motor neurons. Ectodermal co-cultures derived from mouse ES cells formed three-dimensional functional pituitaries following treatment with SAG. In addition, when grafted in hypopituitary mice, these cells secreted adrenocorticotropic hormones and restored appropriate GC levels. A single topical administration of SAG (25 mL, 0.06 mg mL—1) induced the transition to the anagen stage of the hair cycle in adult mouse skin. Treatment resulted in significant upregulation of the Hh target genes GLI1, PTCH1, and GLI2 at the site of administration. In addition, SAG actively upregulated Hh signaling and Gli1 mRNA in cultured human scalp. Treatment of 3T3-L1 cells, a common in vitro model of adipose tissue, with SAG (200 nm) resulted in the rapid (< 5 min) removal of glucose from the culture media with concomitant lactate generation. In vivo, C57BL/6J mice treated with SAG activated the Smo-Ca2+-Ampk axis and stimulated insulin-independent glucose uptake in both muscle and brown fat. Finally, postnatal administration of GCs is a common treatment for pre-term infants that develop chronic lung disease, a treatment that can lead to neurotoxic effects in the cerebellum. Treatment of C57BL/6J mice with SAG (IP injection, 20 mgg—1, once daily for 7 days) provided neuroprotection against GC-derived cerebellar injury and did not interfere with the beneficial lung effects of GC treatment.
Purmorphamine As noted above, C3H10T1/2 mouse embryonic fibroblasts (MEFs) are commonly used as a cellular model of osteoblast differentiation. This differentiation results in the upregulation of alkaline phosphatase (ALP), an enzyme whose activity can be readily assayed in living cells. A high-throughput screen in these cells evaluated approximately 50 000 small-molecule heterocycles to identify inducers of osteogenesis. This screen identified the trisubstituted purine purmorphamine (37) as a potent inducer of osteogenic differentiation in this cell line (EC50 = 1 mm; see Figure 7). Additional in vitro studies demonstrated its ability to induce osteogenesis in pre-osteoblasts (MC3T3-E1 murine cells), and to synergize with BMP-4 to trans differentiate pre-adipocytes (3T3-L1) and myeoblasts (C2C12) into osteoblasts (assays conducted at 1–10 mm). Based on these promising results, a high density oligonucleotide microarray was used to analyze the transcriptional effects of purmorphamine and BMP-4 in C3H10T1/2 cells. Clustering analysis identified a set of genes related to Hh signaling that were uniquely upregulated following purmorphamine treatment (2 mm); specifically, Gli1 (6.9-fold), Gli2 (2.3-fold), Ptch1 (3.3-fold), and Ptch2 (5.9-fold). Purmorphamine upregulated Gli in a dose-dependent fashion (EC50 = 0.5 mm) and its effects were antagonized by multiple Hh pathway inhibitors (Cyc and forskolin), clearly demonstrating that its osteoinductive effects were a direct result of Hh pathway agonism.
A subsequent series of experiments in modified MEFs was used to identify Smo as the cellular target for purmorphamine. Purmorphamine restored Hh pathway activity in Ptch—/— MEFS, yet was inactive in Smo—/— MEFs, indicating it functions downstream of Ptch and suggesting Smo as its cellular target. In addition, purmorphamine displaced BODIPY-Cyc from full-length Smo overexpressed in HEK293T cells (IC50≈ 1.5 mm), suggesting a direct interaction between Smo and purmorphamine. Studies performed in human mesenchymal stem cells (hMSCs) have also demonstrated that purmorphamine treatment (2 mm) activates Hh signaling and initiates differentiation into osteoblasts. While traditional SAR studies based around the purmorphamine scaffold have not been disclosed, a recent report described a ligand-based virtual screen designed to identify structurally related small molecules with similar osteoinductive properties. Three analogues, 38–40, exhibited promising preliminary osteoinductive properties in human mesenchymal stem cells; however, these results did not appear to result from direct activation of Hh signaling.
Purmorphamine has been used in several in vitro systems to explore its therapeutic potential through Hh pathway activation. Dysregulation or degeneration of forebrain GABA interneurons has been implicated as a potential cause of several psychiatric disorders. Purmorphamine was a key component of a chemically defined system that promoted formation of forebrain GABA interneuron development from induced pluripotent stem cell (iPSC)-derived medial ganglionic eminence progenitors. Purmorphamine has also been used as part of a four-step protocol to generate spinal motor neurons from iPSCs. Within the context of cardiovascular regeneration, purmorphamine treatment promoted cardiomyocyte differentiation from hES cell-derived primitive streak cells. Finally, several studies have demonstrated the ability of purmorphamine to commit undifferentiated mesenchymal stem cells into an osteoblast lineage both in vitro and in vivo.
GSA-10
A recent computer-aided drug design effort has identified the quinolinone-based compound GSA-10 (41; see Figure 8) as a new molecular scaffold for developing Smo agonists. The three-dimensional structures of SAG and purmorphamine were used to build a six-feature pharmacophoric model for these two Smo agonists, and this model was subsequently used as the basis for a virtual screen to identify novel molecular structures with the ability to activate Hh signaling. From the in silico screen, 20 “hit” compounds were evaluated for their ability to differentiate the pluripotent mesenchymal cell line C3H10T1/2 into osteoblasts. The most active of these, GSA-10, stimulated differentiation at a level equipotent to purmorphamine (EC50 ≈ 1.2 mm and 0.8 mm, respectively), and while preliminary evidence supported a mechanism for differentiation mediated by canonical Ptch/Smo signaling, follow-up studies provided some interesting discrepancies between Hh activation mediated by SAG or GSA-10. First, treatment with GSA-10 (10 mm) did not upregulate Gli-dependent luciferase activity in NIH3T3 cells and, in fact, it significantly downregulated Gli1 mRNA in C3H10T1/2 cells. GSA-10 did not enhance GCP proliferation in vitro. In addition, it did not induce ciliary accumulation in Smo nor did it compete for Smo binding with BODIPYCyc. Based on this data the authors have suggested that GSA10 stabilizes an as yet unidentified active form of Smo, which does not translocate to the primary cilia; however, further exploration of this compound is needed to fully elucidate its potential mechanism(s) of action.
Summary and Outlook
The Hedgehog (Hh) signaling pathway plays an integral role in correct tissue patterning of a variety of different cell types during embryonic development. In addition, emerging evidence supports a primary role for the Hh pathway in the maintenance and proper differentiation of adult stem cell populations in several cellular contexts. With this in mind, multiple research groups have demonstrated that activation of Hh signaling has potential therapeutic applications in a variety of disease states, including neurodegenerative and osteodegenerative disorders, diabetes, and ischemia. To date, a small number of molecular scaffolds have been identified as Hh agonists; however, extensive SAR studies have only been reported for the biaryl benzothiophenes, characterized by SAG (for Smoothened agonist). SAR reports for the oxysterol scaffold have increased within the last year, and the recent identification of its binding site on the extracellular cystein-rich domain (CRD) of Smoothened (Smo) should provide additional interest into exploring this class of Hh agonists. To date, each compound identified as an Hh agonist has been shown to exert its effects through direct binding and modulation of Smo, a key regulator of Hh signaling and the most “druggable” target within the pathway. Even with promising in vitro and in vivo data supporting activation of Hh signaling as a therapeutic target, several key obstacles must be addressed before Hh pathway agonists advance beyond small molecules of interest into viable drug candidates. First, none of these compounds have advanced past preclinical development and into clinical trials. The most promising class of agonists, the biaryl benzothiophenes under development at Curis, was licensed by Wyeth in early 2004 for extensive preclinical development in models of stroke and cardiovascular injury and by Proctor & Gamble for hair-loss treatment. Both collaborations ended prior to an Hh agonist entering clinical trials, reportedly for safety concerns. While the exact issues encountered for these compounds has not been reported, as many of the disorders targeted by the Hh agonists require chronic administration, long-term safety is a key concern. In addition, as noted above and extensively reviewed elsewhere, constitutive activation of Hh signaling has been implicated in the development of numerous forms of cancer. It is unclear whether the therapeutic effects of prolonged Hh agonism can be separated from the detrimental effects commonly linked to tumor formation; however, the ability to decouple these activities is crucial to moving this class of compounds forward. Finally, mutations in Smo that confer resistance to pathway antagonists and render cancer patients insensitive to treatment with small-molecule Hh inhibitors have been identified.The effects of these mutations on pathway agonism have not been clearly defined; however, the knowledge that mutations can exist within small-molecule binding sites on Smo suggests that this issue must also be addressed for the continued development of Hh pathway agonists.